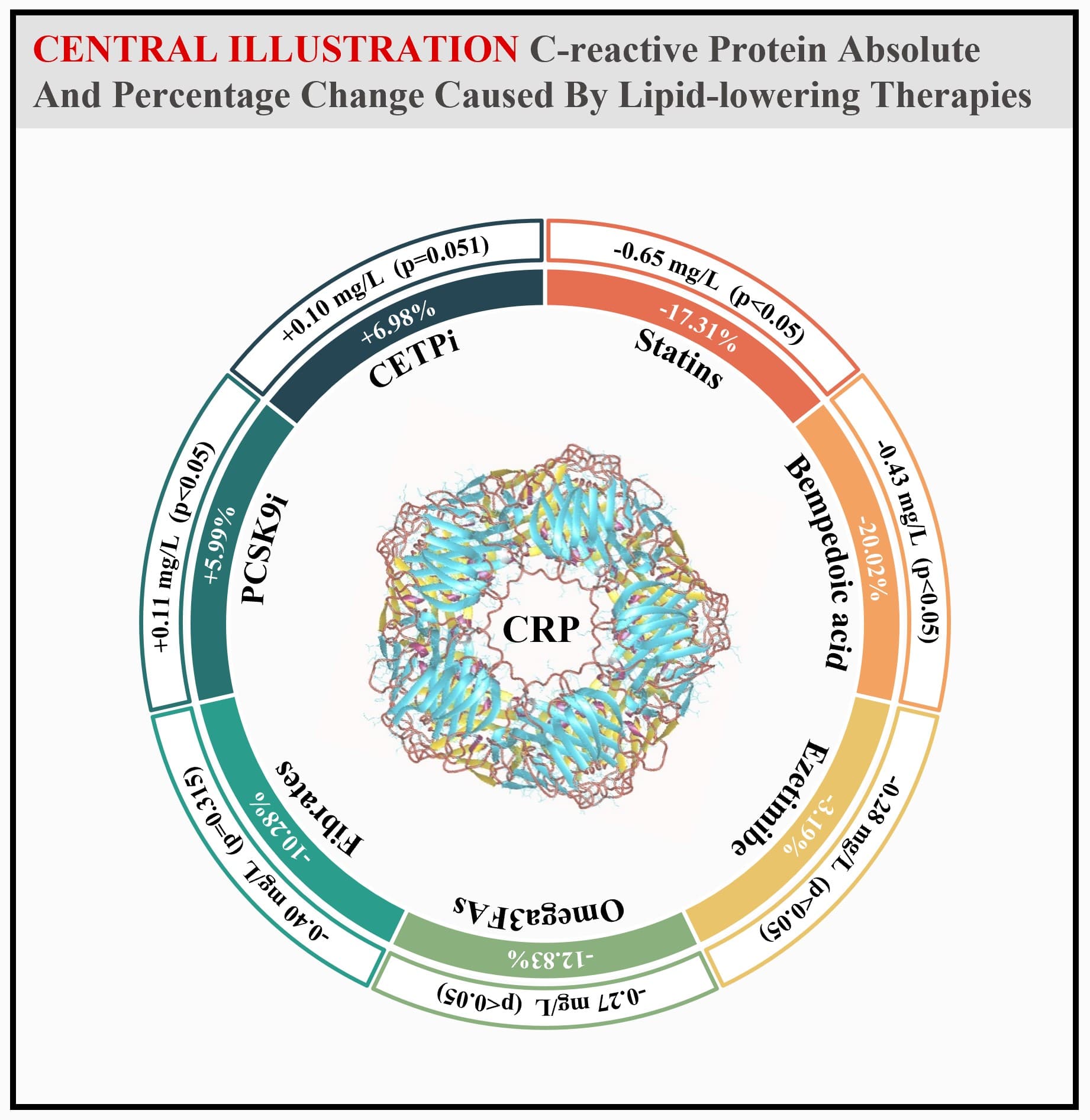
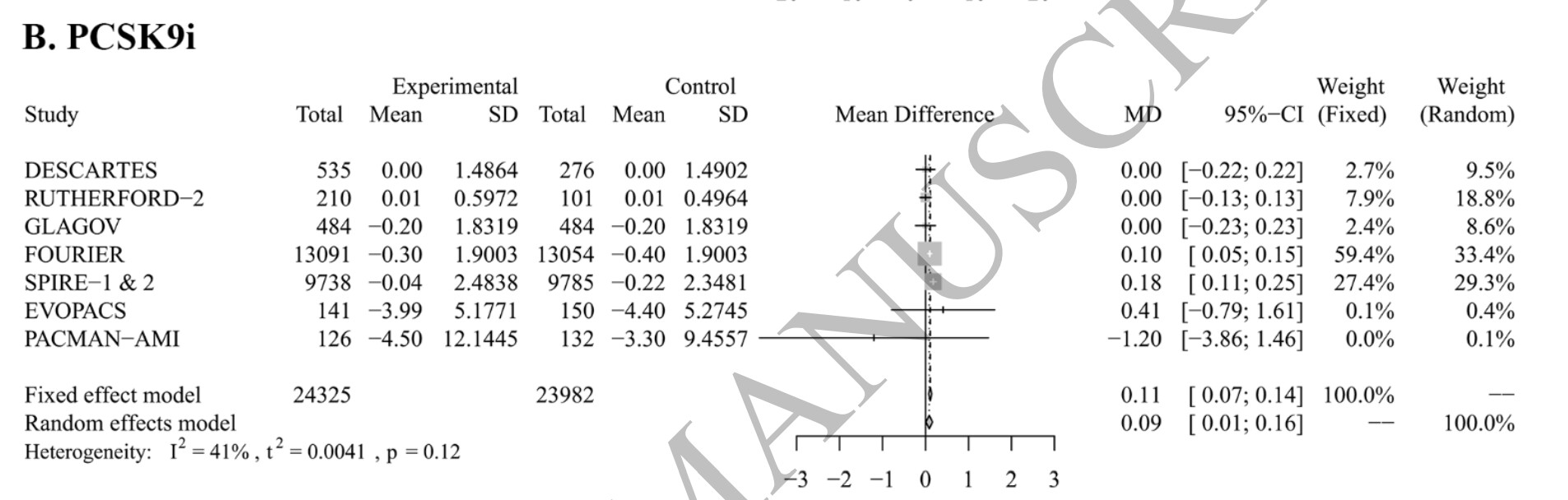
A slight increase of CRP concentration was observed for PCSK9 inhibitors (0.11 [0.07 to 0.14]) and CETP inhibitors (0.10 [0.00 to 0.21]), the latter being not statistically significant.
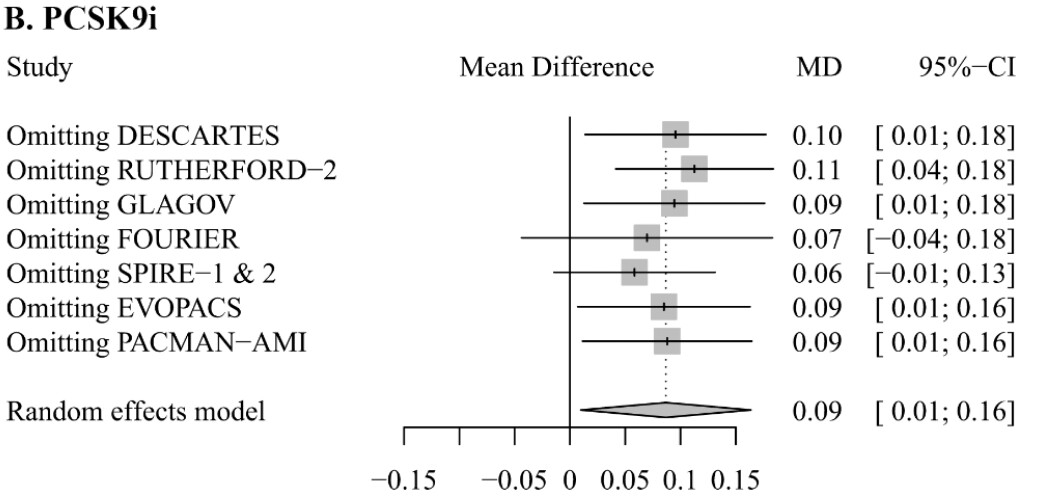
Figure S8: Influence analysis of lipid-lowering therapies on C-reactive protein levels: Forest plots indicating the impact of PCSK9i (B) on C-reactive protein (CRP) levels after omitting each single trial.
in my view there was too much black and whiteness here in what you started out saying is a fact
as you say yourself later in the chain, CETPi was not significant, so it was not optimal to state to the forum that that it increases CRP - do you at least agree with that?
Re
I’m not saying it - it is the authors themselves that are saying it
Here is the part of the paper that covered the effect of PCSK9i on CRP:
When there are two marginally conflicting meta analyses and the authors of the latest one say that their data is confident with the first one, that does not seem to me as the fields underlying has changed in a black and white way.
Anyway, I think the more important questions are
what is the clinical relevance of a 5ish % borderline statistical significant effect from PCSK9i and non-significance effect from CETPi here?
how important are all of the other types of inflammation markers and processes that I shared the paper where PCSK9i might be helpful (and you were silent on/no response to)
is this not a case where N=1 medicine works as it is easy to measure a whole battery of inflammation makers for health optimizers as us here on the forum (and you also were silent on those points)
For me is seems like the jury is still out and not black and white answer for indicate in generally and def not for people who suffer extra inflammation due to Lp(a) - 20% of the population so not a group to generalize our with black and white answers.
I disagree with your reasoning, but… I agree with your conclusion
Indeed, looking in detail at the trials, here are the PCSK9 inhibitors tested in each of the 7 analyzed RCTs:
DESCARTES: evolocumab
RUTHERFORD-2: evolocumab
GLAGOV: evolocumab
FOURIER: evolocumab
SPIRE-1 and SPIRE-2: bococizumab
EVOPACS: evolocumab
PACMAN-AMI: alirocumab
Bococizumab, tested in SPIRE-1 & 2, was withdrawn from development in 2016. The authors note that: “The increase in CRP level caused by PCSK9i became not statistically significant after removing FOURIER (0.07 mg/L [-0.04 to 0.18]) or SPIRE-1 & 2 (0.06 mg/L [-0.01 to 0.13])”
So, we can actually conclude with the authors that PCSK9 inhibition slightly increases CRP however, approved PCSK9 inhibitors are neutral. Still, as the authors noted:
It is worth noting, however, that the design of the included trials for these two treatments frequently containing a run-in phase with statin therapy, which is known to alleviate vascular inflammation. Taken this, both the low CRP levels at baseline and the lack of reduction in this biomarker after treatment could be partially explained, making further investigation necessary.
In any case, it’s easy to measure and adjust if needed.
It is. Do some queries on cortisol and / or glucocorticoids and ASCVD and you’ll get lots of hits…
Cortisol and cardiometabolic disease: a target for advancing health equity
Highlights
Stress in both intrinsic psychosocial and extrinsic physical environmental forms can impact the development of, and outcomes in, cardiovascular disease (CVD).
While experiences of or exposure to stressors may be acute or chronic, its severity and one’s ability to buffer against it is what may be most impactful on the body.
‘Toxic stress’ may affect the body through mechanisms involving the hypothalamic–pituitary–adrenal axis (e.g., cortisol).
Deviations in cortisol diurnal profile have been associated with adiposity, dyslipidemia, incident diabetes, and CVD such as hypertension.
Cortisol and its respective receptor, the glucocorticoid receptor, are involved in metabolism within adipocytes that may contribute to dysglycemia and insulin resistance and, therefore, cardiometabolic disease risk.
Glucocorticoid receptor antagonists and antagonists of the enzymes associated with cortisol metabolism may be promising targets for future research.
Acknowledging and addressing the contribution of stress physiology and cortisol in interventions to treat cardiometabolic disease at the individual, community, and population level are necessary to combat cardiometabolic disease.
Every living human develops senescent cells. However, as you get older, your body (immune system) gets slower and slower at removing them. While a given senescent cell may hang around for 2 or so days in a 20-year-old before being removed, that same cell will take around 20 days to remove for a 60±year-old. As you age, you have more senescent cells because your body can’t remove them as quickly or efficiently as when you were young. Hence the reason for starting senolytics at age 60+ And those senescent cells that hang around may also release SASP causing other normal cells to become senescent.
The solution is senomorphics such as Rapamycin, Taurine and Metformin to prevent senescent cell formation as well as senolytics at a later age (60+) to get rid of any that are hanging around.
Of course, older people develop more senescent cells. Since they aren’t cleaned up as quickly they propagate due to SASP. Therefore more senescent cells develop in older individuals. I’m sure there are other ways as well. This is just one example.
I don’t think either of us have any evidence as to whether the clear up percentage for younger people is higher or lower than older people. If you do I would be interested in reading it.
Which is the one referred to makes the assumption that senescent cells only disappear as a result of the immune system operating on them to kill them. We know that they can also stop being senescent and start functioning. (through senomorphic interventions)
Hence the assumption they base their paper on does not hold.
I think it holds for the majority of people who do not use senomorphics. Of course once you start using senomorphics, the game changes. However I still believe that senomorphics only slow down the accumulation of senescent cells.
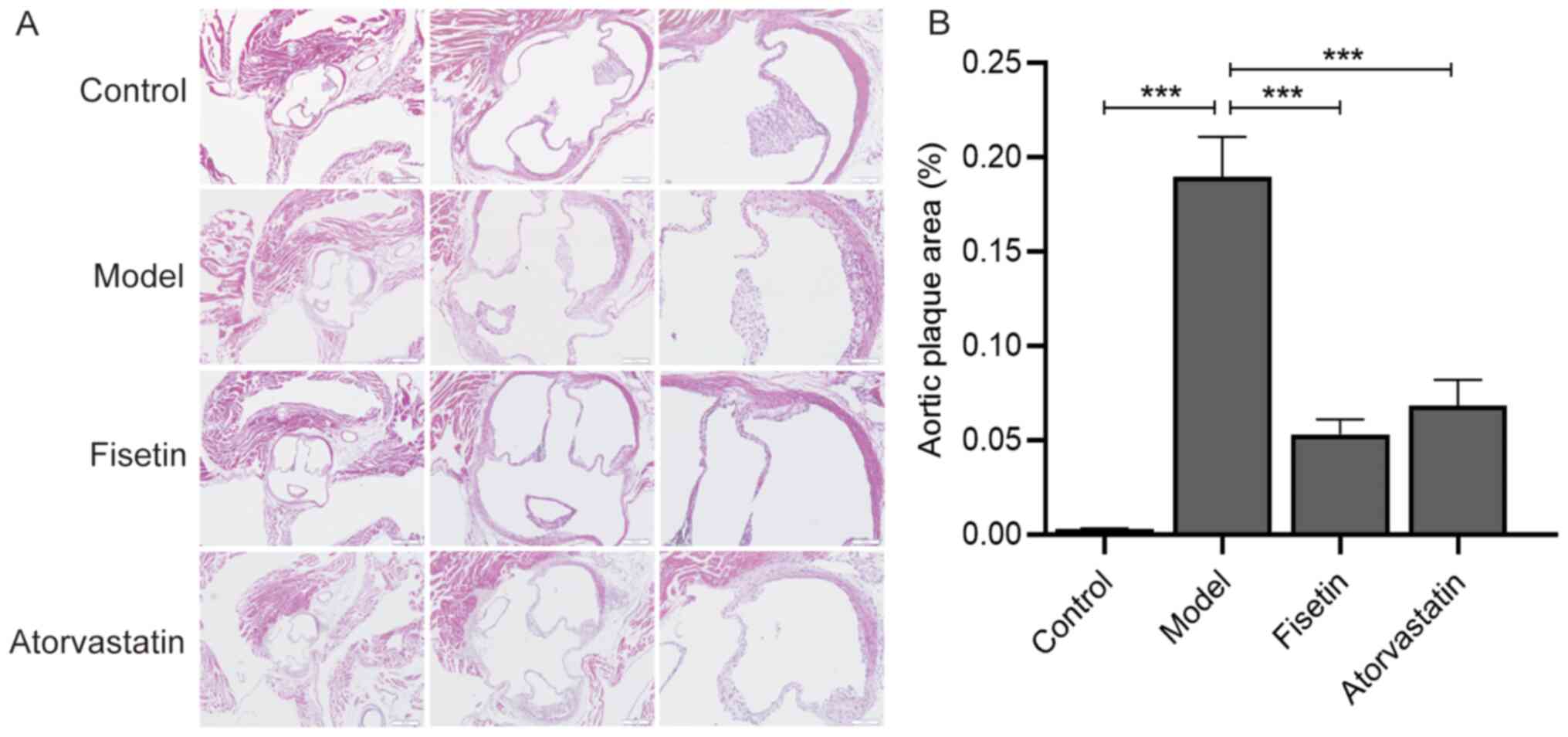
54 apoE-/- 12-week-old mice were fed a normal diet for 1 week before being randomly divided into three groups: apoE-/- mice + high-fat diet (model group; n=18), apoE-/- mice + high-fat diet + fisetin (fisetin group; n=18) and the apoE-/- mice + high-fat diet + atorvastatin (atorvastatin group; n=18).
Mice in the fisetin and atorvastatin groups were gavaged with aqueous solutions of fisetin and atorvastatin, respectively. Via conversion with reference to an adult body weight of 60 kg and the equivalent dose of mice, the final doses of fisetin and atorvastatin aqueous solutions provided to mice were 12.5 and 2 mg/kg, respectively. The fisetin was dissolved in 0.1% DMSO aqueous solution, and the atorvastatin tablets as a positive control drug were directly dissolved in distilled water. The control and model groups were treated with the same volume (0.2 ml/mouse/day) of distilled water via daily oral gavage, and the intervention period of each group was 12 weeks.
Combined with the results of previous in vitro experiments, these results suggested that fisetin may serve an atheroprotective role, ameliorating abnormal lipid metabolism by regulating the expression of PCSK9 and LOX-1, and improving senescence by regulating the expressions of p53, p21 and p16 (Fig. 5).
Dr. Alo seem like he is worth a follow. He explains the mistaken thinking some people have regarding statins. If the PREVENTABLE or STAREE study detect a positive effect on dementia, will everything change? Even if not, it doesn’t prevent someone from taking other drugs or therapies now.